MACRO™
How does MACRO™ work?
1
MACRO™ is BioDynLab’s physics-based Molecular Complexity Analysis platform for peptides and proteins.
During execution MACRO™ processes the trajectory time-histories of all atoms in a given macromolecule. The key output of MACRO™ are the Amino Acid Participation Factors (AAPF).
The AAPFs reflect not only the structure of a macromolecule but also its modes of vibration. They establish a quantitative link between structure, dynamics, information transmission paths and biological function.
The AAPFs link directly to biological properties because they identify amino acids whose dynamic contributions underpin functionality, stability, and interactions with ligands.
What is the input to MACRO™?
2
MACRO™ reads and processes PDB (Protein Data Bank) files which are produced by Molecular Dynamics Simulation codes, both commercial and free tools.
What results does MACRO™ produce?
3
MACRO™ produces Amino Acid Participation Factors, which rank all amino acids in terms of impact on a molecule’s dynamics as well as on the total encoded information. These outputs are produced in a format which makes it easy to integrate MACRO™ into any SW system.
MACRO™ provides a measure of structural robustness which reflects the ability of a protein to withstand perturbations and can affect its half-life.
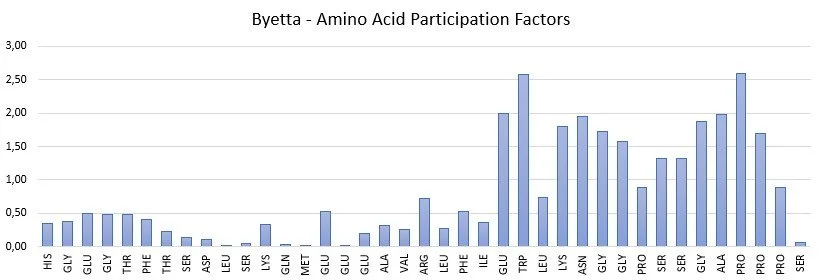
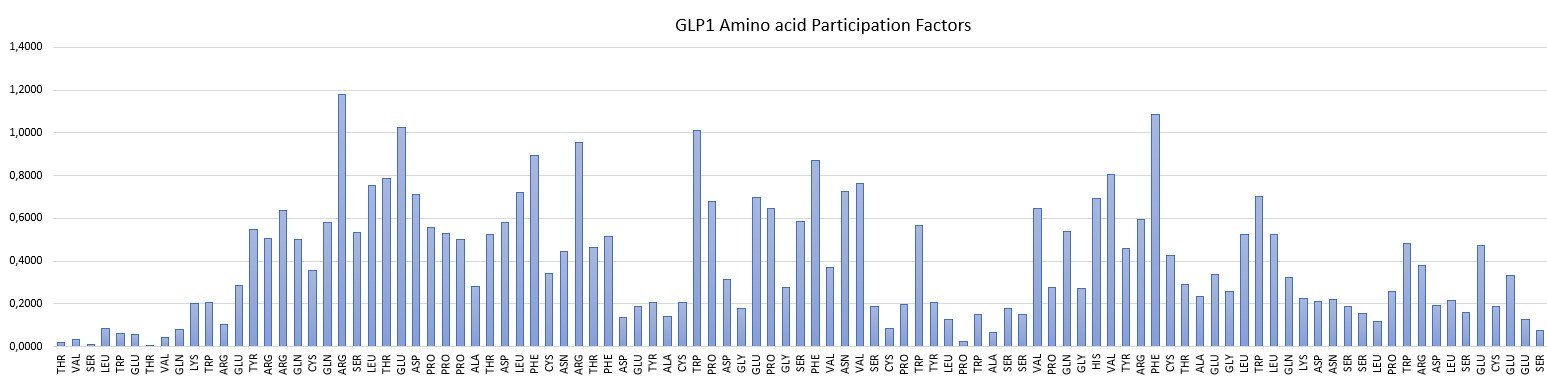
Amino Acid Participation Factors are intrinsic properties of a macromolecule. Examples of AAPF spectra for a peptide and protein are illustrated below.
What is the best computational environment to run MACRO™?
4
MACRO™ runs best on multi-processor Linux systems.